

The increasing implementation of the R programming language in health technology assessments (HTAs) is the result of the need for transparency, reproducibility, and efficiency in health economic modelling. Unlike conventional excel-based tools, R provides a completely script-based setting that records every step of the modelling process, from data import to simulation and reporting. This improves auditability and error reduction while also enabling smooth automation, making R perfectly suitable to cater to the rising demand for “living HTAs” that evolve with new evidence.(1, 2)
HTA bodies, including NICE (the UK) and ZIN (the Netherlands) are adopting R-based submissions, indicating rising institutional confidence in open-source, code-driven methodologies.(3) The ability of R programming manage multifaceted systems, incorporate version control, and automate analyses is changing how HTAs are performed. Academic and industry partnerships are creating shared frameworks and toolkits to further simplify these processes, facilitating consistent, transparent, and faster decision-making.(1, 3)
Standardisation is the most crucial factor of R adoption. For this, validated and reusable modelling frameworks are being developed to help regulators.(4) Initiatives like the open-source assertHE package integrate validation and quality checks right into modelling workflows, supporting built-in verification rather than retrospective review.(5) These frameworks reduce review time, enhance reproducibility, and facilitate efficient model adaptation across markets, thus striking a balance between innovation and rigour. The growing number of health economists equipped with R expertise further reinforces this ecosystem, shifting toward code-based submissions that are easier to review, update, and share.(1, 4, 5)
The move toward standardisation also facilitates scalability in global HTAs. R also facilitates country-specific modifications through modular inputs rather than structural model changes, maintaining consistency across jurisdictions. Shared code sources, scenario templates, and uniform data structures are helpful in cross-country comparisons, making them more reliable and less resource-intensive.(1, 3, 4)
R’s versatility goes beyond modelling efficiency to support real-world data (RWD) and artificial intelligence (AI) integration, which are the crucial pillars of HTA evidence bases. R’s capacity for secure data handling, API-based automation, and remote computation enables models to advance dynamically while maintaining data privacy. However, challenges, especially about data quality, interoperability, and achieving methodological agreement across agencies, persist. Overcoming these warrants collective participation from academia, regulators, and industry to establish shared standards and training guidance.(2, 3, 6)
Finally, the implementation and standardisation of R signify a critical step towards making HTAs more transparent, reproducible, and globally aligned. By adopting open-source technology and collaborative validation, R is transforming the assessment of health technologies, taking HTA from static assessments into dynamic, data-driven systems that adapt to evidence and policy needs of the real-world.
Become A Certified HEOR Professional – Enrol yourself here!
References
- Smith RA, Schneider PP, Mohammed W. Living HTA: Automating Health Economic Evaluation with R. Wellcome Open Res. 2022; 11(7):194.
- R Consortium. R for Health Technology Assessment (HTA): Identifying Needs, Streamlining Processes, Building Bridges. Accessed online on 10th November 2025. Available at: https://r-consortium.org/posts/r-for-health-technology-assessment-hta-identifying-needs-streamlining-processes-building-bridges/
- Poerrier JE, Ettinger J, Bergemann R. R in HEOR modelling for HTA submissions: An assessment. Accessed online on 10th November 2025. Available at: https://www.parexel.com/application/files/2917/2729/8142/FY24_R_in_HEOR_Modelling_White_Paper_09-2024_v3.pdf
- Thokala, P., Srivastava, T., Smith, R. et al. Living Health Technology Assessment: Issues, Challenges and Opportunities. PharmacoEconomics. 2023; 41:227–237.
- Smith RA, Samyshkin Y, Mohammed W, et al. assertHE: an R package to improve quality assurance of HTA models. [version 1; peer review: 1 approved, 1 approved with reservations]. Wellcome Open Res. 2024; 9:701.
- Zisis K, Pavi E, Geitona M, Athanasakis K. Real-world data: a comprehensive literature review on the barriers, challenges, and opportunities associated with their inclusion in the health technology assessment process. J. Pharm. Pharm. Sci. 2024; 27:12302.







 Systematic reviews (SRs) are incredibly crucial for healthcare decision-making, as they often provide a reliable summary of evidence on the comparison among healthcare interventions. They identify, assess, and combine the results of similar but individual studies and help to clarify the known and unknown benefits and risks associated with drugs, devices, and other healthcare interventions. SRs are helpful for clinicians to incorporate research findings into their daily practices, for patients to make informed choices about their care, and for professional medical organizations to develop clinical practice recommendations. (1)
Systematic reviews (SRs) are incredibly crucial for healthcare decision-making, as they often provide a reliable summary of evidence on the comparison among healthcare interventions. They identify, assess, and combine the results of similar but individual studies and help to clarify the known and unknown benefits and risks associated with drugs, devices, and other healthcare interventions. SRs are helpful for clinicians to incorporate research findings into their daily practices, for patients to make informed choices about their care, and for professional medical organizations to develop clinical practice recommendations. (1)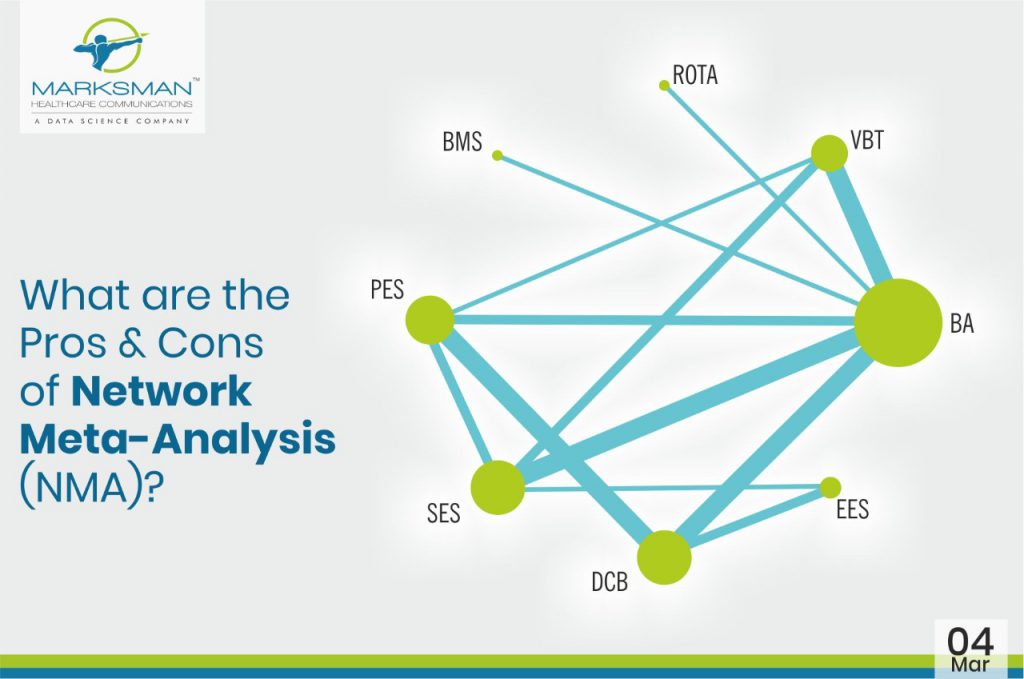
 Evidence-based medicine (EBM) is gaining wide acceptance from researchers globally as it thoroughly optimizes the latest available evidence to make informed care decisions. This involves evaluating the quality of the clinical data by critically assessing methodologies reported in publications. Moreover, EBM incorporates both clinical expertise as well as patient values. Meta-analyses of RCTs often make it among the top of the evidence hierarchy, since it’s regarded as the most valid clinical proof. Indeed, meta-analysis is a validated method to analyse and summarize knowledge by increasing the number of patients, and thus also the effective statistical power. However, there are several limitations associated with meta-analysis, which considers only pairwise comparisons. Unfortunately, head-to-head comparisons are not always available in the literature or they fail to answer a specific clinical question. This can be overcome with the help of network meta-analysis (NMA), which helps providing a global estimate of efficacy or safety of numerous experimental treatments that have not before been directly compared with adequate precision, or at all. Network meta-analysis integrates both direct and indirect effects from the entire set of evidence. Additionally, it ranks the treatments as the best or worst on the basis of valid statistical inference methods. (1)
Evidence-based medicine (EBM) is gaining wide acceptance from researchers globally as it thoroughly optimizes the latest available evidence to make informed care decisions. This involves evaluating the quality of the clinical data by critically assessing methodologies reported in publications. Moreover, EBM incorporates both clinical expertise as well as patient values. Meta-analyses of RCTs often make it among the top of the evidence hierarchy, since it’s regarded as the most valid clinical proof. Indeed, meta-analysis is a validated method to analyse and summarize knowledge by increasing the number of patients, and thus also the effective statistical power. However, there are several limitations associated with meta-analysis, which considers only pairwise comparisons. Unfortunately, head-to-head comparisons are not always available in the literature or they fail to answer a specific clinical question. This can be overcome with the help of network meta-analysis (NMA), which helps providing a global estimate of efficacy or safety of numerous experimental treatments that have not before been directly compared with adequate precision, or at all. Network meta-analysis integrates both direct and indirect effects from the entire set of evidence. Additionally, it ranks the treatments as the best or worst on the basis of valid statistical inference methods. (1)
 Heath Technology Assessment (HTA) is a scientific research area that makes informed clinical as well as policy decisions on the use of health technologies, which include pharmaceuticals, medical devices, diagnostics, procedures and other clinical, public health and organizational interventions. (1)
Heath Technology Assessment (HTA) is a scientific research area that makes informed clinical as well as policy decisions on the use of health technologies, which include pharmaceuticals, medical devices, diagnostics, procedures and other clinical, public health and organizational interventions. (1)
 We all want safe and effective medicines to reach patients as soon as possible, but as we know, drug development, market authorization and payer assessment are all slow sections of a long and drawn out journey for a drug. But what if patients could have access to medicines not just months earlier, but potentially 8 years earlier? This is exactly what the European Medicines Agency (EMA) have in mind, as they lead a broad and diverse group of key stakeholders towards a root-and-branch upheaval of current practice. Adaptive Licensing (AL) (earlier known as adaptive pathways; AP), an ambitious and evolving new initiative which incorporates Real World Evidence (RWE): clinical data collected outside of a conventional randomized controlled trial. AL reforms the existing regulatory approach.
We all want safe and effective medicines to reach patients as soon as possible, but as we know, drug development, market authorization and payer assessment are all slow sections of a long and drawn out journey for a drug. But what if patients could have access to medicines not just months earlier, but potentially 8 years earlier? This is exactly what the European Medicines Agency (EMA) have in mind, as they lead a broad and diverse group of key stakeholders towards a root-and-branch upheaval of current practice. Adaptive Licensing (AL) (earlier known as adaptive pathways; AP), an ambitious and evolving new initiative which incorporates Real World Evidence (RWE): clinical data collected outside of a conventional randomized controlled trial. AL reforms the existing regulatory approach.
 Health systems have developed at different speeds, and with differing degrees of complexity throughout the twentieth century, reflecting the diverse political and social conditions in each country. Notwithstanding their diversity, all systems, however, share a common reason for their existence, namely the improvement of health for their entire populations. To attain this goal a health system undertakes a series of functions, most notably, the financing and delivering of health services.
Health systems have developed at different speeds, and with differing degrees of complexity throughout the twentieth century, reflecting the diverse political and social conditions in each country. Notwithstanding their diversity, all systems, however, share a common reason for their existence, namely the improvement of health for their entire populations. To attain this goal a health system undertakes a series of functions, most notably, the financing and delivering of health services.